
Nano Res.│金属纳米颗粒会在电极表面自发腐蚀?
2021-06-25 16:26:02
作者:研之成理 来源:研之成理
分享至:


背景介绍
探究金属纳米颗粒的氧化还原行为是得以理解它们腐蚀现象的关键。其中的化学变化在诸多领域中都有着重要的启示意义,如水生环境下的纳米毒性学、电子元器件和电池退化问题等。金属纳米颗粒的氧化腐蚀常常伴随着它们的直接或最终溶解,因此,有不少研究在纯液相下探究这些反应。然而,纳米颗粒在盐溶液中极易集聚,这种动态的集聚过程使得剖析此类研究的实验数据变得十分困难。
相比之下,电化学分析方法,如溶出伏安法,一直还是研究此类反应的不二选择,尽管它需要增加一个电极-溶液界面才能够控制反应。可是溶出伏安法必然需要极化电极表面来驱动颗粒的氧化。我们推测,理论上纳米颗粒的氧化腐蚀有些时候也可以在断路电压下被溶液中的活性物质(如溶解氧)驱动。更具体来讲,如图1所示,在这种情况下没有电子流过外部电路,而是电子直接通过电极基底传递到界面上的活性物质;也就是说,电极起到了催化反应并传导电子的作用。本实验就致力于验证这种猜想。
另外,由于一般在研究金属纳米颗粒的氧化还原反应时,我们会假定电极既没有化学活性也没有催化活性,可这并非在所有情况中都成立,甚至有时候电极可能具有决定性的影响。电极本身对此类反应的影响其实是目前纳米颗粒电化学研究领域中容易被忽略的问题。本项研究成果中发现的电极的催化功能为该类科学问题提供了一种回答。

图1氧化还原导致的金属纳米颗粒的溶解反应机理。其中,电子通过宏观尺度的电极基底从金属颗粒转移到界面上任意位置上,很可能相隔很远(远远超过电子隧穿距离),的氧化剂(这里指溶解氧)分子中。
研究方法
在适当条件下,由于其特殊的光学性质(即与光相互作用会导致局域表面等离子共振现象),目前已经可以通过暗视野光学显微镜观察到直径为10-100 nm的银纳米颗粒,即使我们“看到”的是一个个受衍射限制的光斑。因此,这种成像技术已被用来观察溶液中或固定在基底上的金属纳米颗粒。在本项研究中,作者结合非原位光学显微镜(作为主要观察及测量手段)和电化学方法(调控电极电压)来探究上述猜想。
成果简介
作者利用反射暗视野光学显微镜观察到了固定在不同电极基底上银纳米颗粒散射出的光均会随着时间推移慢慢减弱。具体发现这些纳米颗粒在被浸入到了含卤族负离子的溶液中,处于断路电压或者恒电位控制下,其颗粒数量会不断减少。通过结合非原位光学和电化学技术,此现象背后的物理原理被论证为金属纳米颗粒的电化学溶解,其电子被转移到了水中痕量的溶解氧上。作者进一步对比固定在导电性不同的四种基底(即玻璃,铟锡氧化物,玻碳和铂)上的银纳米颗粒的溶解速率。最终发现,所研究的三种导电的基底均可以催化金属纳米颗粒的氧化腐蚀;同样有趣的是,作者推测此过程中电子从金属纳米颗粒通过宏观尺度的电极表面转移到了氧气分子这个电子受体上。
图文导读
要点一:光学显微镜观察到银纳米颗粒在电极表面上的自发消失现象
作者将银纳米颗粒滴镀到一个宏观尺度的玻碳电极表面后干燥,在反射暗视野光学显微镜下可观察到大量的明亮光斑(图2(a)),表示它们的存在。经分析,这些光斑对应的是纳米颗粒的聚合体,而非单个纳米颗粒,因此其散射光强度只能大致表示纳米颗粒的总数。当此表面浸润到0.2 M碘化钾溶液中6分钟后干燥,作者惊奇地发现纳米颗粒的散射光几乎完全消失了,而如果当此表面只浸润到纯水中6分钟后,纳米颗粒依旧大量存在。此外,图2(b)中对应的电极表面也被放置于扫描电子显微镜下观察,确认纳米颗粒本身确实消失了(而不仅仅是变小了或变成散光较弱的其他固体颗粒)。

图2 (a)纳米颗粒被滴镀在电极的局部区域,在暗视野显微镜下的成像。(b)浸润在碘化钾溶液6分钟后的局部电极表面。(c)分别浸润在碘化钾溶液和纯水后,所剩纳米颗粒散射光强度的对比。
要点二:结合光学和电化学方法论证其现象本质以及反应中的氧化剂和还原剂
作者随后将图2(a)中对应的电极表面再度浸润到碘化钾溶液中6分钟,但在此期间施加不同的电极电压,发现在电压足够负时,纳米颗粒的消失几乎可以被完全抑制(图3,黑色散点图)。这个电压阈值(图3,垂直的红色虚线)正好与银纳米颗粒的氧化还原峰位(图3,红色曲线)重合。由此,作者认为,正是因为纳米颗粒的氧化被电极的负电压极化所抑制,我们才可以看到没有消失的银纳米颗粒。这显然表明纳米颗粒的消失是因为它被氧化溶解(或腐蚀)了。

图3 黑色散点为在不同电极电压下浸润于碘化钾溶液6分钟后所剩的银纳米颗粒散射光光强;红色实线为银纳米颗粒在该碘化钾溶液中的氧化还原循环伏安图。虚线为空白电极对照实验结果。
另一方面,通过伏安法测得氩气饱和、空气饱和与氧气饱和的碘化钾溶液中的溶解氧浓度分别为1-2 μM,300 μM和1.2 mM(图4(a))。然而在这三种不同溶解气体环境下的纳米颗粒溶解速率却没有显著变化(图4(b))。并且作者表示常规实验装置很难进一步降低溶解氧的浓度。另外,作者通过计算和实验已经排除了氢离子和碘化钾中微量的碘单质作为此反应的氧化剂。考虑到不论是哪种氧化剂,要使表面浓度低于nM级别的银颗粒完全消失所需的量肯定非常小,并且在要点四中作者推测出氧化剂分子可以在整个电极表面与纳米颗粒发生电子转移,所以推断此反应需要的氧化剂的量极低(远小于1 μM)。综上,作者依然推测此腐蚀反应的氧化剂是溶解氧,并且该氧化反应不是整个反应过程中的决速步骤。
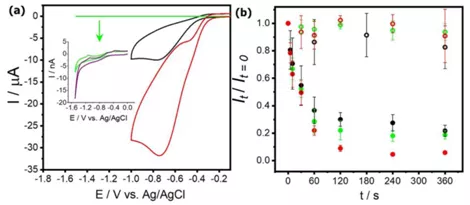
图4 (a)在玻碳电极上空气饱和(黑色)、氧气饱和(红色)的碘化钾溶液中氧还原反应的伏安图,其中插图里对应的碳纤维微电极上氩气饱和(绿色和紫色)溶液气体环境。(b)实心散点分为别浸润于氩气饱和(绿色)、空气饱和(黑色)和氧气饱和(红色)碘化钾溶液6分钟内随时间变化所剩的银纳米颗粒散射光光强。空心散点则为对应的纯水中的对照实验结果。
要点三:光学显微镜下进一步研究银纳米颗粒继氧化后的溶解反应
如果将实验条件中的电解质换成溴化钾、氯化钾或硝酸钾,实验时间调整为50分钟,则可以看到纳米颗粒散射出的光的量在腐蚀实验后有此规律KI<KBr<KCl<KNO3~H2O(图5(a))。此趋势符合可溶性银卤络合物的生成常数的变化趋势。此外,图5(b)表明纳米颗粒的溶解速率随着碘离子浓度的增加而变大。因此,结合之前提到的扫描电子显微镜观察到的纳米颗粒消失的事实,作者推定在碘化钾溶液中银纳米颗粒生成了可溶性银碘络合物,从而完全失去了其散射光。
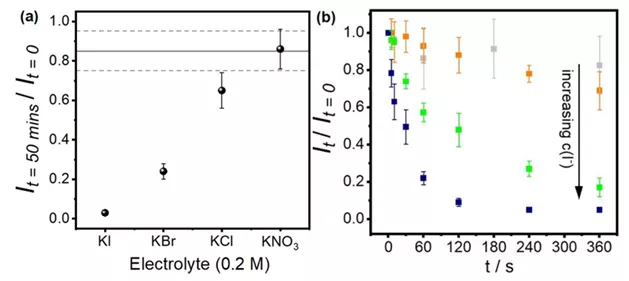
图5 (a)浸润于不同溶液50分钟后所剩的银纳米颗粒散射光光强。水平虚线代表在纯水中浸润得到的结果(b)浸润于不同浓度的碘化钾溶液6分钟内随着时间推移所剩的银纳米颗粒散射光光强。
要点四:(电极)基底在此氧化腐蚀过程的作用与影响

图6 黑色柱状图为在不同基底上滴镀的银纳米颗粒散射光在6分钟浸润于0.2 M碘化钾溶液后所剩光强。白色柱状图为对应的纯水中实验结果。绿色散点为相应基底的电导值。
最后,作者对比了四种不同基底材料上(即玻璃,铟锡氧化物,玻碳和铂,导电性递增;其中玻璃是绝缘体,铂是良好的导体)的纳米颗粒的溶解速率。如图6结果显示,首先,在绝缘体二氧化硅上,溶解依然存在,只是速率很慢。此推论得到了进一步延长反应时间的实验的支持——在大概1.5个小时的浸润之后,纳米颗粒就会完全消失。其次,基底导电性越高,溶解速率越快。于是该实验证明电极表面竟然可以催化此腐蚀溶解反应!
课题组简介
作者牛津大学谢若晨博士,Christopher Batchelor-McAuley博士, 杨民俊博士和Richard G. Compton教授的信息,以及课题组的简介请见http://compton.chem.ox.ac.uk/ (2021年)。
免责声明:本网站所转载的文字、图片与视频资料版权归原创作者所有,如果涉及侵权,请第一时间联系本网删除。
相关文章

官方微信
《中国腐蚀与防护网电子期刊》征订启事
- 投稿联系:编辑部
- 电话:010-62316606-806
- 邮箱:fsfhzy666@163.com
- 中国腐蚀与防护网官方QQ群:140808414
点击排行
PPT新闻

“海洋金属”——钛合金在舰船的
点击数:7130

腐蚀与“海上丝绸之路”
点击数:5741