



(这是一篇写给金属材料人的“内功心法”,请耐心读完)
在高校和科研院所混迹了大半辈子,评审了无数篇论文,也答辩过无数个博士,我发现一个挺尴尬的现象。
你问一个博士生,你的课题研究的是什么?他能滔滔不绝地跟你讲G相、Laves相、拓扑密排相,甚至能把γ‘/γ’相界面的错配度给你算到小数点后两位。但如果你冷不丁地问他一句:“你用的那个‘分子动力学’,它的名词解释到底是什么?它凭什么能算出力来?”
我见过太多人愣住。有的人支支吾吾说是“用电脑算原子轨迹”,有的人则干脆回避,说“这只是个工具,会用就行”。
这种状态,像极了《三体》里那句扎心的话:“你们知道物理,却不理解物理。”
今天,一个研究了一辈子铁碳铬镍的老家伙,不想跟你谈具体的合金成分,只想跟你掰扯掰扯这个所谓的“分子动力学”。如果你愿意沉下心来读这十分钟,我保证,以后你再看到那些花花绿绿的原子模拟图,眼睛里将不再是“动画片”,而是真真切切的“物理规律”。
一、 别再望文生义了,它根本不是什么“分子”动力学
首先,我们要纠偏。
这个翻译害了太多人。“分子动力学”——你一听,以为是研究分子的。但在咱们金属材料领域,我们面对的是什么?是Fe、Ni、Cr、Al这些金属原子组成的晶体,是失去外层电子后的离子实与电子海的结合。
严格来讲,在金属学语境下,它应该叫“原子尺度下的牛顿力学演绎”。
分子动力学(MD)的本质,其实简单得令人发指:它就是解一个方程组。F = m · a,这是牛顿第二定律,三百多年前的产物。MD做的事情,就是把我们面前那块毫米级的镍基单晶高温合金,拆解成一个个遵循牛顿定律的硬球(原子)。
计算机的任务,就是在一飞秒(10^{-15}秒)的时间步长里,做以下循环:
1. 计算当前每一个原子受到周围邻居的合力(F); 2. 根据F=ma,算出此时此刻这个原子的加速度(a); 3. 更新这个原子的速度和位置; 4. 进入下一个时间步,重复一万亿次。
你看,这有什么高深的吗?没有。这甚至没有涉及到量子力学那种“电子要么在这里、要么在那里”的概率云。MD就是经典的、确定性的粒子运动。
那么问题来了:既然原理这么简单,为什么这东西直到最近二十年才在咱们金属界火起来?
答案也很简单:算力不够,还有我们不知道怎么描述“原子间的力”。
二、 真正的灵魂:“势函数”是原子的“性格密码”
刚才说了,第一步是算“合力(F)”。这个F是怎么来的?在宏观世界,弹簧的F=kx,很简单。但在微观世界,两个金属原子隔了一段距离,它们是互相吸引还是排斥?这种相互作用是非常复杂的。
我们不能去解量子力学方程,因为那样计算量太大,一个原子都算不动。所以,我们需要一个经验公式来近似描述原子间的能量关系。这就是势函数。
在金属材料MD领域,最伟大的发明之一,是Daw和Baskes在1984年提出的嵌入原子法(EAM,Embedded-Atom Method) [1]。
这个理论特别有意思。它认为,一个金属原子在晶体里的能量,由两部分组成:
• 第一部分是“对势”,就像两个磁铁靠近时的斥力或引力; • 第二部分是“嵌入能”,也就是这个原子钻到周围金属电子云“海洋”里游泳时,需要付出多大的能量代价。
这恰恰抓住了金属键的本质:它不是两个原子之间的单纯拉拽,而是所有原子共享的自由电子气在起作用。
有了EAM这个框架,我们才能给不同的合金系统“定制”势函数。比如,为了研究Inconel 718的钎焊扩散,Bonny等人开发的Fe-Ni-Cr势函数,必须能精准地再现不同温度下Ni、Fe、Cr的自扩散系数[2]。如果这个势函数不准,算出来的“扩散”就会变成水在油里扩散,完全违背物理定律。
所以,如果你问我什么是分子动力学,我会说:它是给每一个金属原子赋予了“拟人化”的性格,让它们在虚拟的时空里,按照既定的物理规则(势函数)去碰撞、扩散、滑移、断裂的一场大型沉浸式戏剧。
三、 我们到底在“看”什么?从位错到裂纹的视觉盛宴
我们金属材料人最关心什么?变形。
宏观上,我们看应力-应变曲线;微观上,我们做EBSD看取向差;在纳米尺度,我们看TEM照片里的黑线——那是位错。
但在MD的世界里,位错不再是照片上的“黑线”,而是一条真实在运动的“线缺陷”。
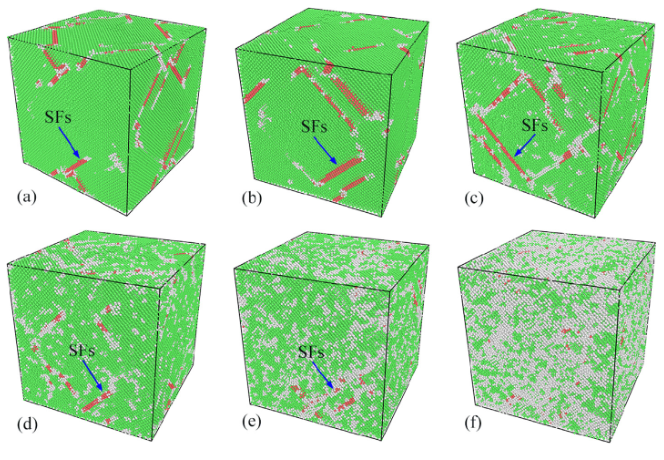
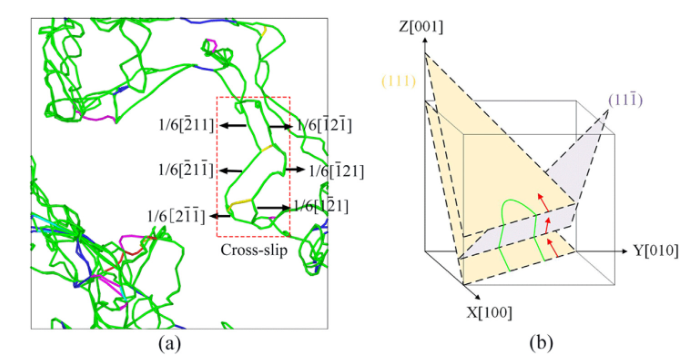
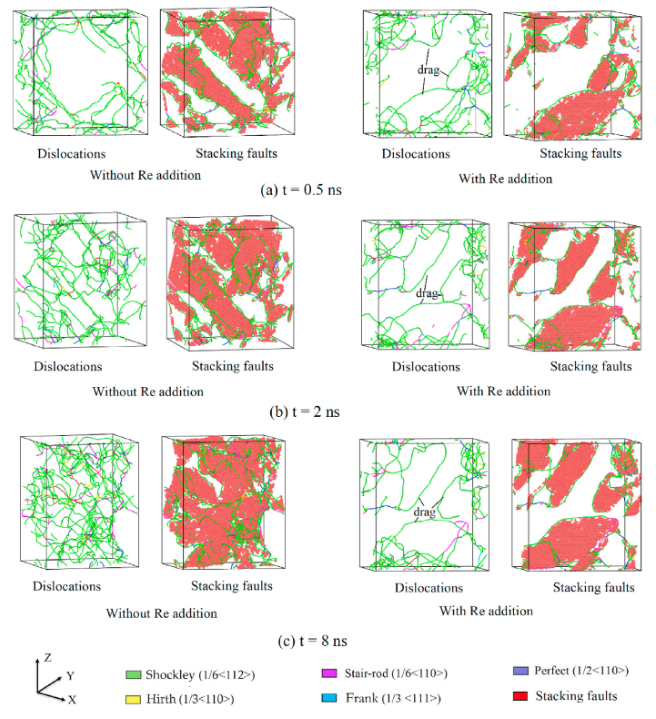
举个例子,最近的研究热点:含Re(铼)与不含Re的镍基单晶高温合金,为什么蠕变寿命差了几十倍?实验上,我们只能通过断口分析推测Re拖曳了位错。但在MD里,Wu等人的模拟直接把这一过程可视化了出来[3]。
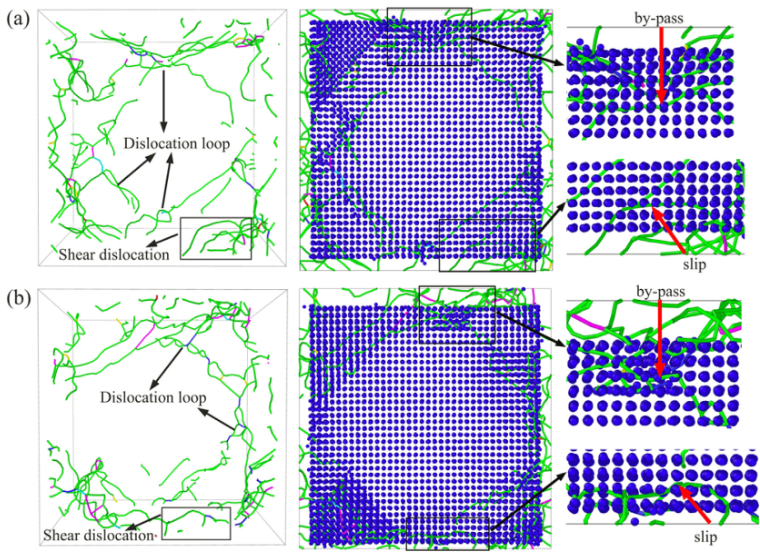
当你拉伸温度达到1200K时,你会看到:γ基体中的位错线像一根橡皮筋一样,在γ‘强化相面前被挡住,随着应力增大,这根橡皮筋弯曲成半圆——这就是经典的Orowan绕过机制。而当你在合金里加入Re原子后,你会看到这根位错线在绕过沉淀相时,总是“黏糊糊”的,因为Re原子偏聚在位错核心,增加了位错攀移所需的激活能。
这不是猜想,这是“看”到的事实。
再比如疲劳。我们做低周疲劳实验,往往要做几百个小时,最后看到裂纹萌生了。但Chen等人的MD模拟[4],在几纳秒内就复现了这一过程。他们发现,在300K低温下,位错是“切”过γ‘相的,留下大量的层错;而在1000K高温下,位错是“绕”过去的。这个机制的转变,直接对应了宏观上疲劳寿命的突变点。
这就是MD的魅力:它把宏观的力学性能(强度、寿命),硬生生地还原为了原子尺度的时间-空间演化。
四、 打破砂锅问到底:MD的“阿喀琉斯之踵”
讲了这么多,你是不是觉得MD是万能的?如果你这么想,那你离真正的“理解”还有一步之遥。
一个真正懂MD的金属材料博士,不仅要知道它能干什么,更要知道它不能干什么。
第一,时间尺度的诅咒。
MD的时间步长是飞秒(fs)。为什么?因为原子的振动频率就是这么快,步子再大一点,原子就飞出去了。这就导致,MD模拟一“帧”能跑的时间,通常只有纳秒(ns)到微秒(μs)级别。
而现实中的蠕变断裂,可能是几百个小时。这就好比,你用每秒24帧的高速摄影去拍一棵树的生长,你拍到了树叶在风中颤抖(原子振动),但你永远拍不到树干变粗的过程(晶粒长大、扩散蠕变)。
所以,当你看到有人用MD模拟“淬火”过程时,你要知道,他的淬火速率是10^9 K/s,比现实快了百万倍。这不是造假,这是受限于算力的“不得已而为之”。 我们必须承认,MD看到的往往是“趋势”和“机理”,而不是绝对的“工程数值”。
第二,尺度的困境。
现在的MD极限,大约能做到亿级原子,大概是几百纳米见方。而在一个毫米级的晶粒里,有成千上万条位错。MD看到的,往往是“单打独斗”的一两条位错与界面的相互作用。那些位错间的缠结、割阶、以及群体效应,MD目前还无能为力。
这也就是为什么,现在顶级的科研都在做“多尺度模拟”——MD算原子细节,把结果提炼成参数,交给晶体塑性有限元(CPFEM)去算宏观构件的响应[5]。
五、 结论:什么是分子动力学?(一个金属学人的终极回答)
文章写到这里,如果你再问我那个原问题:“分子动力学的名词解释是什么?”
我会放下手中的保温杯,认真地告诉你:
分子动力学,不是一种简单的软件操作,而是一种“还原论”视角下的科学信仰。它相信,金属材料宏观上表现的强度、塑性、韧性、疲劳、蠕变,这些看似复杂甚至模糊的工程概念,本质上都可以被还原为几十万个原子在飞秒尺度下,遵循牛顿定律运动的总和。
它是在计算机里构建的一座“虚拟金属实验室”。在这个实验室里,我们可以随心所欲地改变温度(从0K到2000K),改变加载方式(拉伸、剪切、疲劳),甚至像上帝一样在晶格里“点出”一个空位或注入一个Re原子,然后静静观察这个微观宇宙如何演化。
它是连接“量子力学”与“宏观力学”的一座桥梁。量子力学告诉我们原子为什么结合(势函数的来源),宏观力学告诉我们零件什么时候断(应力-应变),而MD告诉我们:那个断裂的过程,究竟是原子们经历了怎样的挣扎、塞积、绕过和撕裂。
最后,我想对年轻的材料学子们说一句:别把MD当黑箱,也别把它当万能药。去读一读那篇经典的EAM原始论文,去手算一下最简单的Lennard-Jones势下的二体运动。只有当你明白了每一个力是怎么来的,每一个速度是怎么更新的,你再看那些花花绿绿的模拟图时,你看到的将不再是色彩,而是物理本身。
到那时,如果有人再问你“什么是分子动力学”,你可以微微一笑,告诉他:
“那是我们窥探金属灵魂的窗口。”
参考文献
[1] Daw, M. S., & Baskes, M. I. (1984). Embedded-atom method: Derivation and application to impurities, surfaces, and other defects in metals. Physical Review B, 29(12), 6443. (金属原子尺度模拟的奠基之作,提出了EAM势函数)
[2] Bonny, G., Castin, N., & Terentyev, D. (2013). Interatomic potential for Fe–Ni–Cr alloys. Modelling and Simulation in Materials Science and Engineering, 21(8), 085004. (用于镍基合金扩散行为模拟的关键势函数开发工作)
[3] Wu, W., Chen, B., Shen, H., & Ding, Z. (2022). Molecular dynamics simulation of rhenium effects on creep behavior of Ni-based single crystal superalloys. Progress in Natural Science: Materials International, 32(2), 259-266. (从原子尺度揭示了Re元素钉扎位错、提升蠕变抗性的机理)
[4] Chen, B., Wu, W. P., Chen, M. X., & Guo, Y. F. (2020). Molecular dynamics study of fatigue mechanical properties and microstructural evolution of Ni-based single crystal superalloys under cyclic loading. Computational Materials Science, 185, 109954. (系统研究了温度对镍基单晶合金疲劳变形机制转变的影响)
[5] Roters, F., et al. (2019). DAMASK – The Düsseldorf Advanced Material Simulation Kit for studying crystal plasticity. Computational Materials Science, 158, 420-478. (目前主流的晶体塑性模拟平台,常与MD进行多尺度耦合)
[6] Plimpton, S. (1995). Fast parallel algorithms for short-range molecular dynamics. Journal of Computational Physics, 117(1), 1-19. (LAMMPS软件的经典原始文献,大多数金属MD模拟的代码基础)
免责声明:本网站所转载的文字、图片与视频资料版权归原创作者所有,如果涉及侵权,请第一时间联系本网删除。

官方微信
《腐蚀与防护网电子期刊》征订启事
- 投稿联系:编辑部
- 电话:010-62316606
- 邮箱:fsfhzy666@163.com
- 腐蚀与防护网官方QQ群:140808415

“海洋金属”——钛合金在舰船的

腐蚀与“海上丝绸之路”